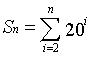
Written by Dr. Árpád Furka, university professor
Budapest, May 29, 1982
As
exemplified, among others, by the peptide hormones discovered so
far, the shorter-lenghtier peptides take part in a number of important
functions in the living organism. It can be supposed, that only a small
fraction is known of these biologically active peptides having potential
therapeutic effect. This fact motivates the intensive international and
domestic research activity in this field.
Two,
in principle different, approaches offer themselves for searching for peptides
bearing new biological effects:
Nn = 20n
If the n-residue peptides are synthesized stepwisely and independently, the number of the require synthetic steps (Sn) can be calculated as follows:
Sn = (n-1) 20n
It is noted, that a synthetic step means
a complete coupling cycle, that is, in addition to the coupling step itself
incorporates the operations connected with the protecting groups, too.
With
good organization, that is, choosing a systematic synthesis route the number
of synthetic steps can be reduced. The minimum number of synthetic steps
is:
The synthesized peptides are supposed to be submitted to screening tests. Since several tests have to be done on each peptide, the total number of the required screening tests is hopelessly large. If the number of kinds of screening tests is denoted by t, the total number of screening tests is expressed by the following equation:
Tn = t 20n
Table
1 shows the the possible number of peptides depending on the number of
residues, the number of synthetic steps required for their synthesis, and
number of the screening tests, calculating with 10 different tests (t=10).
The figures - which are rounded - clearly show, that even the synthesis
and testing of all tripeptides would be an almost hopeless venture.
Table 1
Possible number of peptides (Nn
) containing different number of residues (n),
the number of synthetic steps required for their
synthesis (Sn) in an optimized
process, furthermore the number of screening
experiments (Tn) calculating
with 10 different screening tests (t=10)
(the figures are rounded)
|
n
|
|
|
|
|||
|
2
|
4
|
hundred |
4
|
hundred |
4
|
thousand |
|
3
|
8
|
thousand |
8
|
thousand |
80
|
thousand |
|
4
|
160
|
thousand |
168
|
thousand |
2
|
million |
|
5
|
3
|
million |
3
|
million |
30
|
million |
|
6
|
64
|
million |
67
|
million |
640
|
million |
|
7
|
1
|
billion |
1
|
billion |
13
|
billion |
|
8
|
25
|
billion |
26
|
billion |
256
|
billion |
|
9
|
512
|
billion |
537
|
billion |
5
|
trillion |
|
10
|
10
|
trillion |
10
|
trillion |
102
|
trillion |
Because of the very large number of possible peptides, the stepwise synthesis of all peptides - even in the case of small ones - is an unrealizable task. The large number of the screening experiments constitutes a further problem. The proposal to be outlined on the next pages will try to somewhat improve this almost hopeless situation.
Systematic search for biologically active small peptides through synthesis and screening of peptide mixtures
The proposal to be outlined here constitutes a
research project which makes possible to search for biologically active
peptides with much greater chance than before. When I write down this project
I'm fully aware of its potential importance in industry. It is also clear,
that it's realization is possible only through cooperation of different
institutions. Primarily the participation of the pharmaceutical industry
is desirable since the investments can be recovered through pharmaceutical
industry.
The essence of the proposal is that instead
of one by one synthesis of peptides, peptide mixtures should be prepared
containing several hundred or several thousand peptides in approximately
1 to 1 molar ratio, and these peptide mixtures should be submitted to screening
tests. It will be shown that on this way much labor can be saved both in
the synthetic work and in the screening experiments. In the first stage
one has to determine whether or not the mixture shows any biological effect.
If biological effect is observed, of course, it has to be determined which
component (or which components) are responsible for the activity.
Method for synthesis of peptide mixtures
Since not single peptides but rather mixtures of peptides are synthesized, post synthetic purification and removal of by-products are out of question. Because of this, the classical method of synthesis (in solution) can not be used either. In the synthesis of peptide mixtures the solid phase method have to be applied. It is noted here, that in the syntheses not necessarily the 20 amino acids are used. In some cases more than 20 amino acids may be used, for example if - in addition - non-common amino acids are intended to be used as building blocks. Less than 20 amino acids may be used, for example, in decapeptides, since the synthesis of all peptides seems to be unrealistic and have to compromise with the use of fewer kinds of amino acids. Let denote by k the number of the amino acids intended to vary in the i position. The numbers of amino acids varied in the C-terminal and N-terminal position are k1 and kn , respectively.
Realization of the synthesis. The resin
is divided into k1 equal portions (that is to as
many portions as many amino acids are intended to vary at the C-terminal
of peptides). Then each portion of resin is coupled with one of the
k1 kinds of amino acids then the amino-protecting
group is removed from every sample. A small quantity is removed from every
sample and they are taken aside for later use, then the samples are thoroughly
mixed. Then the mixture of aminoacyl resins is divided into k2
equal portions and each of them is coupled with one of the k2
kinds of protected amino acids then the amino-protecting groups are removed
from each sample. Before mixing, again small samples are removed and taken
aside. The mixture of dipeptides is cleaved from a small portion of the
mixed resin to use it in biological tests. The rest of the mixed resin
is divided into k3 equal parts and the amino acids
intended to occupy the third position are coupled to them. Then the synthesis
is likewise continued until the mixture of n-residue peptides is
reached.
It
is worthwhile to add some notes. As in an ordinary solid phase synthesis,
one has to make an effort to achieve good conversion by applying the reagents
in excess. Fortunately, however, conversions lower than 100%, or minor
unwanted splitting reactions do not cause so serious problems like in ordinary
syntheses. The labour requirement could be significantly reduced by using
mixtures of properly protected amino acids in acylation reactions. This,
however, does not seem to be an acceptable solution because of the differences
in the reactivity of the activated amino acids which would lead to the
formation of peptides in significantly different concentrations thus causing
problems in the screening experiments. Formation of peptides in equal concentrations
can only be assured by mechanical mixing of samples followed by dividing
into equal portions. This makes possible a complete conversion for every
amino acid component. Possibility of acylations with mixtures of several
amino acids of identical reactivity might be a matter of further considerations.
Smaller differences in reactivities could be compensated by properly selected
molar ratios of the amino acid derivatives of the mixture. In the following
calculations the possibility of acylations with the mixtures of amino acid
derivatives will be left out of considerations.
The
number of peptides formed in the synthesis, that is, the number of components
in the peptide mixtures - in a general case - can be calculated by the
following formula:
Nn = k1.k2 . . . . . . kn-1.kn
If the same number (k) of amino acids are varied in every position
Nn = kn
The number of synthetic steps in the synthesis of a peptide mixture containing Nn peptides (considering the attachment of the first amino acid to the resin as separate step) is:
Sn = k1 + k2 + . . . . + kn-1 + kn
If the same number (k) of amino acids are varied in each position,
Sn = nk
The formulae show the advantage of the synthesis
of peptide mixtures: the number of the synthetic steps can be calculated
by summing the numbers of the varied amino acids, while the number peptides
is given by the product of the numbers of the varied amino acids.
One
example: synthesis of the mixture of tetrapeptides prepared by varying
the 20 kinds of amino acids, needs only 80 synthetic steps! It is noted,
that in the same run all shorter peptides - that is the 400 dipeptides
and the 8000 tripeptides - are formed, too. The traditional synthesis of
these peptides would need 168 400 synthetic steps. A different comparison:
in the traditional method with 80 steps only about 30 tetrapeptides can
be synthesized.
Screening of peptide mixtures
Peptides mixtures - in the first approximation - are synthesized to determine whether or not they contain biologically active component. It is supposed - although it needs experimental verification - that screening experiments can be made with mixtures, too. This offers great advantage over the traditional method since the number of screening tests is reduced by a factor equal to the number of components of the mixture. For example, the mixture of the 8000 tripeptides can be examined by a single series of tests. If there is active peptide among them, one of the executable t tests gives positive result. If the number of active peptides is more than one, then, of course, more tests may give positive result. In the synthesis of the mixture of n-residue peptides it is wortwhile to test the shorter peptides, too. The synthesis is so designed to allow for this. Taking this requirement into account, and the number of kinds of tests being t, the total number of the executable tests is:
Tn = t(n-1)
Although this equation certainly holds, its realizability in practice deserves some notes. There is - without any doubt - an upper limit in the number of components of the peptide mixtures to be submitted to screening tests. It is difficult to estimate this number without experiments. The mixtures may probably contain many thousands of components, and as it can be judged today, the method outlined above is rather limited by possibilities of screening tests than by the number of the required synthetic steps. If there are too many components in the mixture, too large samples have to be applied in the screening experiments to achieve observable effect for a single component. The mixture supposedly contains a number of more or less active analogs and their effect is probably summarized. Nevertheless, an unsurpassable limit in the number of components certainly exists. Therefore in certain cases may prove useful to examine the effect of the n-residue mixtures without final mixing. In other cases the synthesis should be designed so not to surpass the optimal number of components.
"Backsearching" for the active peptide. If the peptide mixture is detected to contain active component, that is, if the mixture shows a new type biological effect, then the further task is the isolation and structure determination of the active peptide followed by its synthesis. Once the mixture containing the active component or components is in our hand the isolation can be carried out using the effective separation methods, since these make possible to separate the active compound even from thousands of inactive components. It is possible, however, to follow a different method, too. This will be outlined here. This approach to the identification of the active peptides is supposed to be less tedious then the isolation method, moreover it supplies additional information concerning the structure-effect relationship. Applicability of the method requires a procedure for quantitative determination of activity. For the sake of simplicity let's suppose that the mixture contains a single effective component (besides analogs having the same kind of effect but smaller activity).
Backsearching step No. 1. The experiments are started with the kn samples taken aside in the synthesis of the n-residue peptides before final mixing. The mixtures of n-residue peptides are cleaved from each resin sample. The mixtures of peptides differ from each other in the n-th (that is the N-terminal) residue of their component peptides. Each peptide mixture is submitted to a quantitative activity determination. This shows how the activity depends on the terminal amino acid residue, that is, this way we can determine the N-terminal residue of the active peptide, and in addition it will show the effect of its replacement by other amino acid residues. Let's suppose, for example, that the N-terminal residue in the sample showing the highest activity (as well as in the active peptide) is Phe (phenylalanine). It is noted here that if there are several samples showing equally high activity it is practical to choose as the N-terminal residue of the active peptide the cheapest or the synthetically less problematical amino acid. This note holds for the subsequent backsearching steps, too.
Backsearching step No. 2. The experiment is continued with the kn-1 samples taken aside in the synthetic stage of the (n-1)-residue peptides. The amino acid determined before, that is Phe in our example, is coupled to each sample. Cleavage of the peptides from the support gives kn-1 different peptide mixtures. Their common feature is that every peptide has Phe in the N-terminal position. By submitting the peptide mixtures to quantitative screening experiments one can determine the amino acid residue occupying position n-1 (that is, the pre-N-terminal position) in the active peptide. This experiment also shows the effect on activity of substitution of this amino acids with other ones. Let's suppose that the pre-aminoterminal amino acid is Arg (arginine). It should be noted that in this backsearching step the Phe is coupled to kn-1 samples and the same number (kn-1) of screening experiments have to be done. Not all of the t kinds of tests are required, only the one proved before to be positive. Consequently the number of the synthetic steps and the number of screening experiments are the same: kn-1. It is also noted that in the previous backsearching step only screening test are done (their number is kn) synthetic steps are not needed.
Backsearching step No. 3. This, and the subsequent backsearching steps may be realized using two different approaches. The peptides in samples taken aside during the synthesis have to be elongated to contain n residues, in such way, to carry on their N-terminal section the amino acid residues assuring activity. This can be realized on two ways. Either by stepwise coupling with amino acids (in our example with protected Arg then Phe) or by coupling in a single step with a previously synthesized oligopeptide having the required sequence (in our example Phe.Arg). The required synthetic steps in the two approaches significantly differ. The number of the screening experiments, however, are the same in both cases. Let's turn now to the No. 3. backsearching step.
Stepwise elongation. Let's take the kn-2 samples taken aside in the synthesis of (n-2)-residue peptides. Each sample is coupled first with protected Arg then with protected Phe. After cleaving the peptides from the support each of the kn-2 peptide mixtures are submitted to activity tests to determine the amino acid residue occupying the third position counting from the N-terminal end. The number of screening tests to be executed is kn-2. The number of the required synthetic steps is: 2kn-2. The multiplying factor preceding k is the bigger the shorter are the peptides to be elongated. The numerical value of the factor is equal to the number of amino acids to be coupled with in the elongation process.
Elongation with oligopeptide. A previously synthesized dipeptide (in our example Phe.Arg) is coupled to each of the kn-2 samples taken aside, then the process is continued as described above. The number of screening test is also kn-2. The number of synthetic steps (leaving out of consideration the synthesis of the oligopeptide) is also kn-2. This procedure seems to be more economical. In practice it means that the active peptide is synthesized in parallel with the screening tests using the classical method started from the N-terminus. Small fractions of the growing peptide are sacrificed in the backsearching steps. This backsearching method has the great advantage (in addition to the fact that it needs less synthetic steps) that when the backsearching procedure is finished the active peptide is synthesized, too.
Backsearching
of more than one active peptide. In the synthetic peptide mixtures
several active peptides may be present, showing different effects. In these
cases the number of backsearching steps will be bigger by a factor equal
to the number of the differing active peptides. That is, if the number
of the active peptides is "a" the values deduced above are multiplied
by a. It is noted that the presence in the mixture of peptides having
different effects may complicate the backsearching process especially in
the case of peptides with opposing effects. This, however, is not treated
in details.
The
backsearching process ends when the sequences of all active peptides are
determined by applying either the oligopeptide or the stepwise elongation
method.
Total number of synthetic steps and screening tests summarized
for the whole synthetic backsearching process
Number of synthetic steps using oligopeptide elongation
In synthesis:
![]()
![]()
Total in synthesis and backsearching:
![]()
If k amino acids are varied in each step:
Sn = [n(a + 1) - a]k
Number of synthetic steps using stepwise elongation.
In synthesis:
![]()
![]()
![]()
![]()
If k amino acids are varied in each step:
![]()
Number of screening tests equally valid using the oligopeptide and stepwise elongation
In synthesis: Tn = t(n-1)
In backsearching:
Total in synthesis and backsearching:
![]()
If k amino acids are varied in each step:
Tn = t(n-1) + ank
An example: preparation and screening of all pentapeptides
N5 = 320000, n=5 k=20 t=10 a=1
Total number of synthetic steps
Ogopeptide elongation
180
Stepwise elongation
300
Number of tests
140
Extension of the method to other types of compounds
Applicability of the method outlined before is not restricted for only the systematic searching for active peptides. The same principle applies to all other sequential types of compounds, that is, when the compounds belonging to this type of compounds differ from each other only in their building blocks or the sequences of these building blocks. Among them may occur natural compounds like oligosaccharides or oligonucleotides but synthetic products may be taken into account, too. Among these later ones one may think about sequential copolymers or sequential polycondensates.
Dr. Arpad Furka
university professor
File number 36237/1982
I certify this stitched document comprising 14,
that is, fourteen pages was subscribed in my presence by Dr. Arpad Furka,
university professor, with his own hands.
Budapest, 1982. Nineteen hundred and eighty two,
June 15, (fifteen).
Dr. Judit Bokai
state notary public
Back to Main Page